
本節課程,需要完成擴增子分析解讀1質控 實驗設計 雙端序列合併 先看一下擴增子分析的整體流程,從下向上逐層分析 分析前準備 # 進入工作目錄 cd example_PE250 上一節回顧:我們拿到了雙端數據,進行了質控、並對實驗設計進行了填寫和檢查、最後將雙端數據合併為單個文件進行下游分析。 接下來 ...
本節課程,需要完成擴增子分析解讀1質控 實驗設計 雙端序列合併 先看一下擴增子分析的整體流程,從下向上逐層分析
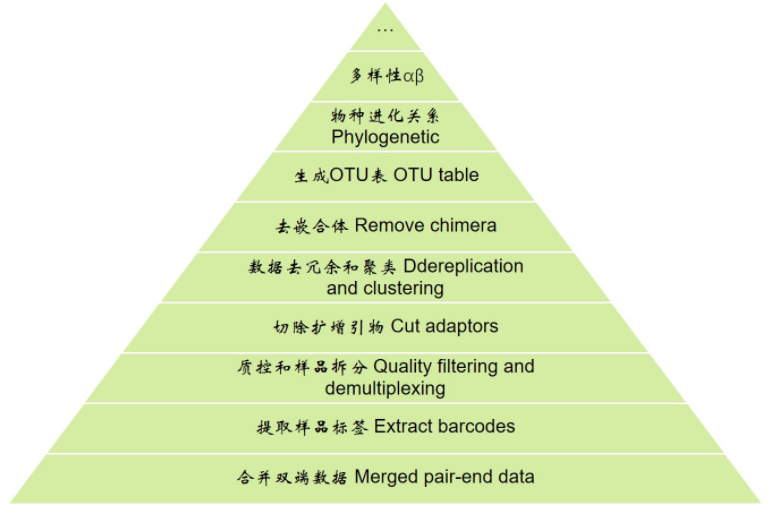
# 進入工作目錄 cd example_PE250上一節回顧:我們拿到了雙端數據,進行了質控、並對實驗設計進行了填寫和檢查、最後將雙端數據合併為單個文件進行下游分析。 接下來我們將序列末端的barcode標簽切下來,因為它們是人為添加的,不屬於實驗對象;再根據標簽序列與實驗設計文件比對,對每條序列屬於哪個樣品進行分類;最後我們切除掉擴增使用的引物,因為它們是人工合成的相似序列,並不是物種的真實序列。這樣我們就獲得了高質量的擴增區域數據,並且序列名稱中包括了樣品信息。 4. 提取barcode 為什麼擴增子有barcode? 我以前做過sRNA-Seq, RNA-Seq, ChIP-Seq等等,都是一個文庫對應一個樣品,為什麼沒有用過barcode拆分。 原因是擴增子目前研究對象細菌、真菌多樣性沒有表達基因數量大,一般是幾百到千的水平,對數據量要求最多10萬條序列即可飽合。而Illumina測序儀的通量很高,採用Index來區分每個文庫,每個文庫的數據量達到千萬的級別,加上建庫測序的成本也不會低於千元。對於擴增子動輒成百上千的樣品既太貴,又浪費。因此將擴增子樣本添加上barcode(標簽),通常將48/60個樣品混合在一起,構建一個測序文庫,達到高通量測序大量樣品同時降低實驗成本的目的。 通常的測序儀下機數據,只經過Index比對,拆分成來自不同文庫的數據文件,分發給用戶。而擴增子的一個文庫包括幾十個樣品,還需要通過每個樣品上標記的特異Barcode進一步區分,再進行下游分析。 註:如果你是自己構建測序文庫,返回數據可以按下文拆分樣品;如果是公司建庫並測序,他們有樣品的barcode信息,通常會返給用戶樣品拆分後的數據,無需本節中的操作。但原理還是要理解,對整體分析的把握非常重要。 Barcode在擴增子的位置和類型?
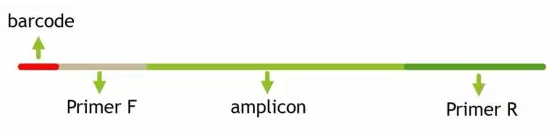 Barcode位於引物的外側,比較典型的有三種,上圖展示的為最常用的barcode位於左端(正向引物上游),此外還有右端和雙端也比較常用。
本分析採用的數據類型為右端barcode。
extract_barcodes.py是QIIME中用於切除barcode的腳本,支持你想到的所有類型。
-f參數為輸入文件,即上文中合併雙端數據後的文件;
-m為實驗設計文件;
-o為輸出切下barcode的數據目錄;
-c為barcode類型選擇,目前的barcode_paired_stitched選項支持右端和雙端類型,如果是左端類型,請改為barcode_single_end;
—bc1_len設置左端barcode的長度,按實驗設計添寫,本數據只有右端則為零;
—bc2_len設置右端barcode的長度,按實驗設計添寫,本數據右端長度為6bp,常用的還有8,10;
-a是使用實驗設計中的引物來調整序列的方向,本實驗的測序無方向,必須開此選項調整,有些公司的建庫類型有方向,但開了總沒錯,頂多多花點計算時間;
—rev_comp_bc2是將右端barcode取反向互補再與左端相連,建議打開,這樣你的實驗設計中barcode一欄直接將添寫原始barcode即可,不用再考慮方向了;如果是雙端則將兩個barcode直接連起來填在barcode列,不用考慮方向。
Barcode位於引物的外側,比較典型的有三種,上圖展示的為最常用的barcode位於左端(正向引物上游),此外還有右端和雙端也比較常用。
本分析採用的數據類型為右端barcode。
extract_barcodes.py是QIIME中用於切除barcode的腳本,支持你想到的所有類型。
-f參數為輸入文件,即上文中合併雙端數據後的文件;
-m為實驗設計文件;
-o為輸出切下barcode的數據目錄;
-c為barcode類型選擇,目前的barcode_paired_stitched選項支持右端和雙端類型,如果是左端類型,請改為barcode_single_end;
—bc1_len設置左端barcode的長度,按實驗設計添寫,本數據只有右端則為零;
—bc2_len設置右端barcode的長度,按實驗設計添寫,本數據右端長度為6bp,常用的還有8,10;
-a是使用實驗設計中的引物來調整序列的方向,本實驗的測序無方向,必須開此選項調整,有些公司的建庫類型有方向,但開了總沒錯,頂多多花點計算時間;
—rev_comp_bc2是將右端barcode取反向互補再與左端相連,建議打開,這樣你的實驗設計中barcode一欄直接將添寫原始barcode即可,不用再考慮方向了;如果是雙端則將兩個barcode直接連起來填在barcode列,不用考慮方向。
# 提取barcode extract_barcodes.py -f temp/PE250_join/fastqjoin.join.fastq \ -m mappingfile.txt \ -o temp/PE250_barcode \ -c barcode_paired_stitched --bc1_len 0 --bc2_len 6 -a --rev_comp_bc2這步任務會在輸出目錄temp/PE250_barcode中生成5個文件
barcodes.fastq # 切下來的barcode,用於後續拆分樣品 barcodes_not_oriented.fastq # 方向不確定序列的barcode。連引物都不匹配,質量太差,建議不再使用 reads1_not_oriented.fastq # 方向不確定序列的序列,可能barcode切錯方向。連引物都不匹配,質量太差,不建議使用 reads2_not_oriented.fastq # 空文件 reads.fastq # 序列文件,與barcode對應,用於下游分析更多說明建議閱讀幫助 http://qiime.org/scripts/extract_barcodes.html 5. 質控及樣品拆分 上步對序列方向進行了調整全部為正向,並切開了barcode與擴增序列。下麵使用split_libraries_fastq.py對混池根據barcode拆分樣品,同時篩選質量大於20(即準確度99%)的序列進行下游分析。參數解釋如下: -i 輸入序列文件,上步產生; -b 輸入barcode文件,上步產生; -m 實驗設計,依賴樣品barcode列表將序列按樣品重新命名 -q 測序質量閾值,20為99%準確率,30為99.9%準確 --max_bad_run_length 允許連續的低質量鹼基調用的最大值,預設值為3 --min_per_read_length_fraction 最小高質量序列比例,0.75即最少有連序75%的鹼基質量高於20 --max_barcode_errors barcode 允許的鹼基錯配個數,建議設置0為不允許,即使修改為1,2通常也不允許錯配,不信你試試 barcode_type 調置barcode類型,預設為golay_12,並支持錯配;我們通常設置為整數,對應barcode的長度總和,本實驗中填0+6=6。
# 質控及樣品拆分 split_libraries_fastq.py -i temp/PE250_barcode/reads.fastq \ -b temp/PE250_barcode/barcodes.fastq \ -m mappingfile.txt \ -o temp/PE250_split/ \ -q 20 --max_bad_run_length 3 --min_per_read_length_fraction 0.75 --max_barcode_errors 0 --barcode_type 6運行結果會輸出三個文件
histograms.txt # 所有序列長度分佈數據,可知長度範圍308-488,峰值為408 seqs.fna # 質控並拆分後的數據,序列按樣品編號為SampleID_0/1/2/3 split_library_log.txt # 日誌文件,有基本統計信息和每個樣品的數據量;查看可知每個樣品最大數據量為110454,最小值為10189更多說明建議閱讀幫助http://qiime.org/scripts/split_libraries_fastq.html 6. 切除引物序列 這裡我們介紹一款高通量引物切除軟體,cutadapt,2017-6-16最新版1.14; https://pypi.python.org/pypi/cutadapt 此軟體2011年發佈至今一直在更新,Google Scholar截止17年8月8日引用2263次。 Cutadapt 1.14下載和安裝
# 下載,請儘量檢查主頁下載最新版源碼 wget https://pypi.python.org/packages/16/e3/06b45eea35359833e7c6fac824b604f1551c2fc7ba0f2bd318d8dd883eb9/cutadapt-1.14.tar.gz # 解壓 tar xvzf cutadapt-1.14.tar.gz # 進入程式目錄 cd cutadapt-1.14/ # 安裝在當前用戶目錄,不需管理員許可權 python setup.py install --usercutadapt切除雙端引物及長度控制,參數詳解: -g 5’端引物 -a 3’端引物,需要將引物取反向互補 -e 引物匹配允許錯誤率,我調置0.15,一般引物20bp長允許3個錯配,為了儘量把引物切乾凈 -m 最小序列長度,根據情況設置,本實驗擴增V5-V7區,長度主要位於350-400,故去除長度小於300bp的序列,無經驗可不填此參數 --discard-untrimmed 引物未切掉的序列直接丟棄,防止出現假OTU seqs.fna 為輸入文件 PE250_P5.fa 為輸出文件
cutadapt -g AACMGGATTAGATACCCKG -a GGAAGGTGGGGATGACGT -e 0.15 -m 300 --discard-untrimmed temp/PE250_split/seqs.fna -o temp/PE250_P5.fa程式運行結果會在屏幕上輸出結果統計報告,如去接頭比例、去掉過短序列比例等;還有去除引物的長度分佈,看看有沒有異常。如3’端序列沒有取反向互補會無法去除19bp序列,而是幾bp的錯誤序列。 Cutadapt結果報告
This is cutadapt 1.14 with Python 3.6.1
Command line parameters: -g AACMGGATTAGATACCCKG -a GGAAGGTGGGGATGACGT -e 0.15 -m 300 --discard-untrimmed temp/PE250_split/seqs.fna -o temp/PE250_P5.fa
Trimming 2 adapters with at most 15.0% errors in single-end mode ...
Finished in 73.83 s (58 us/read; 1.04 M reads/minute).=== Summary ===
Total reads processed: 1,277,436
Reads with adapters: 1,277,194 (100.0%)
Reads that were too short: 8,849 (0.7%)
Reads written (passing filters): 1,268,345 (99.3%)Total basepairs processed: 522,379,897 bp
Total written (filtered): 495,607,409 bp (94.9%)=== Adapter 1 ===
Sequence: GGAAGGTGGGGATGACGT; Type: regular 3'; Length: 18; Trimmed: 202757 times.
No. of allowed errors:
0-5 bp: 0; 6-12 bp: 1; 13-18 bp: 2Bases preceding removed adapters:
A: 96.3%
C: 1.5%
G: 0.8%
T: 1.3%
none/other: 0.0%
WARNING:
The adapter is preceded by "A" extremely often.
The provided adapter sequence may be incomplete.
To fix the problem, add "A" to the beginning of the adapter sequence.Overview of removed sequences
length count expect max.err error counts
3 3 19959.9 0 3
4 4 4990.0 0 4
6 2 311.9 0 2
8 1 19.5 1 1
11 1 0.3 1 1
13 1 0.0 1 1
15 9 0.0 2 9
17 42 0.0 2 42
18 202626 0.0 2 202626
19 56 0.0 2 56
20 1 0.0 2 1
21 1 0.0 2 1
32 1 0.0 2 1
38 1 0.0 2 1
39 1 0.0 2 1
41 1 0.0 2 1
309 2 0.0 2 2
310 1 0.0 2 1
311 3 0.0 2 3=== Adapter 2 ===
Sequence: AACMGGATTAGATACCCKG; Type: regular 5'; Length: 19; Trimmed: 1074437 times.
No. of allowed errors:
0-5 bp: 0; 6-12 bp: 1; 13-19 bp: 2Overview of removed sequences
length count expect max.err error counts
3 2 19959.9 0 2
7 1 78.0 1 0 1
8 2 19.5 1 1 1
10 6 1.2 1 4 2
11 1 0.3 1 1
12 3 0.1 1 2 1
13 5 0.0 1 3 2
14 24 0.0 2 17 7
15 51 0.0 2 32 14 5
16 71 0.0 2 56 12 3
17 134 0.0 2 92 30 12
18 327 0.0 2 189 117 21
19 1059175 0.0 2 1056863 2069 243
20 13846 0.0 2 1817 10955 1074
21 744 0.0 2 5 10 729
22 1 0.0 2 1
23 2 0.0 2 2
24 1 0.0 2 1
25 2 0.0 2 2
27 5 0.0 2 5
28 2 0.0 2 2
29 2 0.0 2 2
30 1 0.0 2 1
31 2 0.0 2 2
32 10 0.0 2 10
49 1 0.0 2 1
51 1 0.0 2 1
166 1 0.0 2 1
291 6 0.0 2 6
401 2 0.0 2 2
409 1 0.0 2 1
443 1 0.0 2 1
460 2 0.0 2 2
465 2 0.0 2 2WARNING:
One or more of your adapter sequences may be incomplete.
Please see the detailed output above.

