
在使用FastQC之後,如果我們發現了一些問題(序列質量不高),那麼我們該使用什麼樣的工具,去解決這些問題呢? fastx Toolkit是包含處理fastq/fasta文件的一系列的工具,它是基於java開發的,我們高通量測序最常用到的是使用這個軟體進行reads的裁剪(trim) FASTQ-t ...
在使用FastQC之後,如果我們發現了一些問題(序列質量不高),那麼我們該使用什麼樣的工具,去解決這些問題呢? fastx Toolkit是包含處理fastq/fasta文件的一系列的工具,它是基於java開發的,我們高通量測序最常用到的是使用這個軟體進行reads的裁剪(trim) FASTQ-to-FASTA 說明:這個命令主要是用來轉換FASTA格式與FASTQ格式
fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i] [-o][-h] = 獲得幫助信息 [-r] = 使用序號去代替fastq文件中原來的reads名 [-n] = 如果fastq中有N,保留(預設是有N的序列刪除) [-v] = 報告reads的總數 [-z] = 調用GZip軟體,輸出的文件自動經過壓縮 [-i] = 輸入文件,可以是fastq/fasta格式 [-o] =輸出路徑,如果不設置會直接輸出到屏幕 FASTX Statistics 說明:主要統計一下序列的基本信息,如GC含量什麼的,很少用,基本使用FastQC代替
fastx_quality_stats [-h] [-i INFILE] [-o OUTFILE][-h] = 獲得幫助信息 [-i] = FASTA/Q格式的輸入文件 [-o] = 輸出路徑,如果不設置會直接輸出到屏幕 FASTA/Q Clipper 說明:主要是進行reads的過濾和adapter的裁剪
fastx_clipper [-h] [-a ADAPTER] [-i INFILE] [-o OUTFILE][-h] = 獲得幫助信息 [-a ADAPTER] = Adapter序列信息,預設的是CCTTAAGG [-l N] = 如果1條reads小於N就拋棄,預設5 [-d N] = 保留adapter並保留後面的Nbp,如果設置-d 0等於沒有用這個參數 [-c] = 只保留包含adapter的序列 [-C] = 只保留不包含adapter的序列 [-k] = 報告adater的序列信息 [-n] = 如果reads中有N,保留reads(預設是有N的序列刪除) [-v] = 報告序列總數 [-z] = 調用GZip軟體,輸出的文件自動經過壓縮 [-D]= Debug output FASTA/Q Trimmer 說明:這個是我最常用的工具,可以快速切序列
fastx_trimmer [-h] [-f N] [-l N] [-z] [-v] [-i INFILE] [-o OUTFILE][-h] = 獲得幫助信息 [-f N] = 序列中從第幾個鹼基開始保留,預設是1 [-l N] = 序列最後保留到多少個鹼基,預設是整條序列全部保留 [-z] = 調用GZip軟體,輸出的文件自動經過壓縮 cutadapt軟體 這個cutadapt軟體是最常用的去adapter的工具。它是基於Python編寫的一個Python包 一些最基本的用法 # cutadapt的功能特別強大,相對應的參數真的特別多,有幾十個參數,我們平時只會用到很少的幾個,我在這裡為大家介紹一下。 # 最基本的形式,可以去掉3‘端的adapter序列
cutadapt -a AACCGGTT -o output.fastq input.fastq# 可以直接輸入或者輸出壓縮文件,不需要修改參數,輸出文件的後面加上.gz
cutadapt -a AACCGGTT -o output.fastq.gz input.fastq.gz# 假如去掉3‘端的adapter AAAAAAA 和5’端的adapter TTTTTTT
cutadapt -a AAAAAAA -g TTTTTTT -o output.fastq input.fastq# cutadapt也可以用來進行reads的cut,去掉最前面的5bp
cutadapt -u 5 -o trimmed.fastq input_reads.fastq# 進行reads測序質量的過濾 # cutadapt軟體可以使用-q參數進行reads質量的過濾。基本原理就是,一般reads頭和尾會因為測序儀狀態或者是反應時間的問題造成測序質量差,比較粗略的一個過濾辦法就是-q進行過濾。需要特別說明的是,這裡的-q對應的數字和phred值是不一樣的,它是軟體根據一定的演算法計算出來的 # 3‘端進行一個簡單的過濾,--quality-base=33是指序列使用的是phred33計分系統
cutadapt -q 10 --quality-base=33 -o output.fastq input.fastq# 3‘端 5’端都進行過濾,3'的閾值是10,5‘的閾值是15
cutadapt -q 10,15 --quality-base=33 -o output.fastq input.fastqReads 長度的過濾 [--minimum-length N or -m N] # 當序列長度小於N的時候,reads扔掉 [--too-short-output FILE] # 上面參數獲得的這些序列不是直接扔掉,而是輸出到一個文件中 [--maximum-length N or -M N] # 當序列長度大於N的時候,reads扔掉 [--too-long-output FILE] # 上面參數獲得的這些序列不是直接扔掉,而是輸出到一個文件中 Paired-Reads的裁剪(trim) # 現在很多的測序都是雙端測序,那麼從測序原理上來說,一對reads來自於1簇反應,所以一起進行adapter的trim可能效果更好。cutadapt自然也提供了這樣的功能
cutadapt -a ADAPTER_FWD -A ADAPTER_REV -o out.1.fastq -p out.2.fastq reads.1.fastq reads.2.fastq# -a是第1個文件的adapter序列 # -A是第2個文件的adapter序列 # -o是第1個輸出文件 # -p是第2個輸出文件 1個例子 其實在實際中,我們從公司拿到的數據大多數已經進行過cutadapt,我們其實更關註的是reads的trim 我們首先要用fastqc對test1.fastq與test2.fastq進行一下質量評估,評估的主要結果如下: read1的測序質量圖
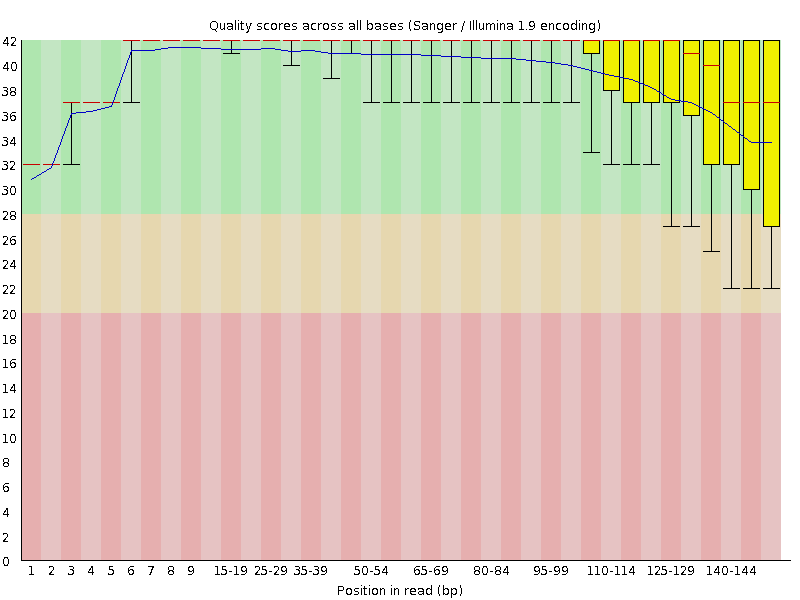 read2的測序質量圖
read2的測序質量圖
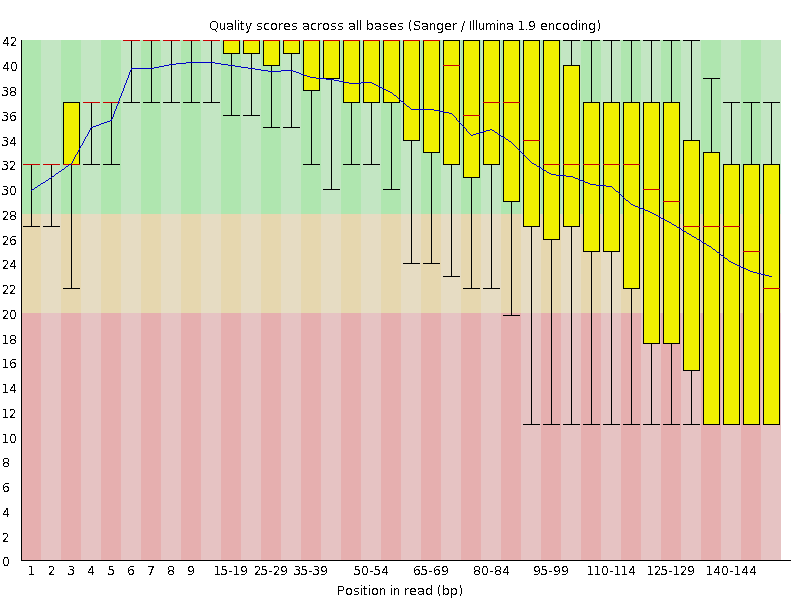 我們從上面兩張圖可以明顯看出,read1的測序質量明顯好於read2,一般我們確定要trim多少bp的時候都是按phred20這個標準進行評估。比如,對於我們測試數據,read1就不需要trim,read2需要保留1-85bp。對應的fastx_trimmer的命令如下:
我們從上面兩張圖可以明顯看出,read1的測序質量明顯好於read2,一般我們確定要trim多少bp的時候都是按phred20這個標準進行評估。比如,對於我們測試數據,read1就不需要trim,read2需要保留1-85bp。對應的fastx_trimmer的命令如下:
fastx_trimmer -i test_data_2.fastq -o test_data_2_trim.fastq -f 1 -l 85[-f N] = 序列中從第幾個鹼基開始保留,預設是1 [-l N] = 序列最後保留到多少個鹼基,預設是整條序列全部保留

